|
|
|
This webpage aims to provide some hand-on exercises for the course of "Computational Design of Materials". Procedures of model preparation and ab initio calculation of material properties are illustrated in details. I compiled seven case studies for the hand-on workshops. These case studies represent only my personal interest. I have gone through every step to make sure it works. More examples will be included in the near future. I hope these examples can be modified easily to fit to your needs. |
Tutorial on Quantum EspressoIn this section, I would like to describe the procedures that I used to conduct first-principles simulation on the electronic structures and material properties and gain hand-on experiences of quantum espresso. The newest version of QE will be used. You can refer to my webpage for how to install the package and the libraries needed. Here I use silicon and aluminum to illustrate the simulation procedures on semiconductor and metal, respectively. You can download the structural files from ICSD or any other public domain database and build the crystal structure with xcrysden, VESTA or any crystal builder. After we have the unit cell information, the unit cell structure of a crystalline material is always relaxed and then self consistent calculation (scf) is completed. After that, we can estimate the elastic modulus of the crystals and calculate its band structure. Along this line, we will analyze the density of states to learn what atomic orbitals contribute significantly to the occupied and unoccupied bands. Semiconductor: SiliconA. Structural Relaxation
A structural relaxation can be done by specifying the 'calculation' flag with either 'relax' or 'vc-relax' (vc meaning variable cell). The difference between the two is that in 'vc-relax' the cell parameter itself is allowed to change, whereas in 'relax' the cell is fixed but the atoms can move within the cell. You can prepare an input file to run quantum espresso module pw.x. To save your effort, you can download a shell script "Si-vcrelax.sh" here to automate the process. Run the shell script as follows: B. SCF Run
1. Prepare an input file for silicon scf run with calculation = 'scf' and celldm(1) = 10.31335 C. Equation of State (EoS)
1. Prepare the shell script volume.sh 
FIG.1: Equation of state (EoS) of silicon single crystal. D. Charge Density Plot on a Specific Plane
1. Prepare a shell script run-si.scf.sh for scf calculation. Execute the code with 
FIG.2: Charge density plot on (110) plane of silicon crystal. E. Conduct a nscf Run for Analyzing Density of State
1. Prepare a shell script run-si.nscf.sh for nscf calculation on 6x6x6 k-mesh. Execute the code with 
FIG.3: Density of state (DoS) plot of silicon crystal. The vertical line at 5.93 eV marks the highest occupied level.
4. Producing Partial DoS plot with run-si.pdos.sh and execute the code. 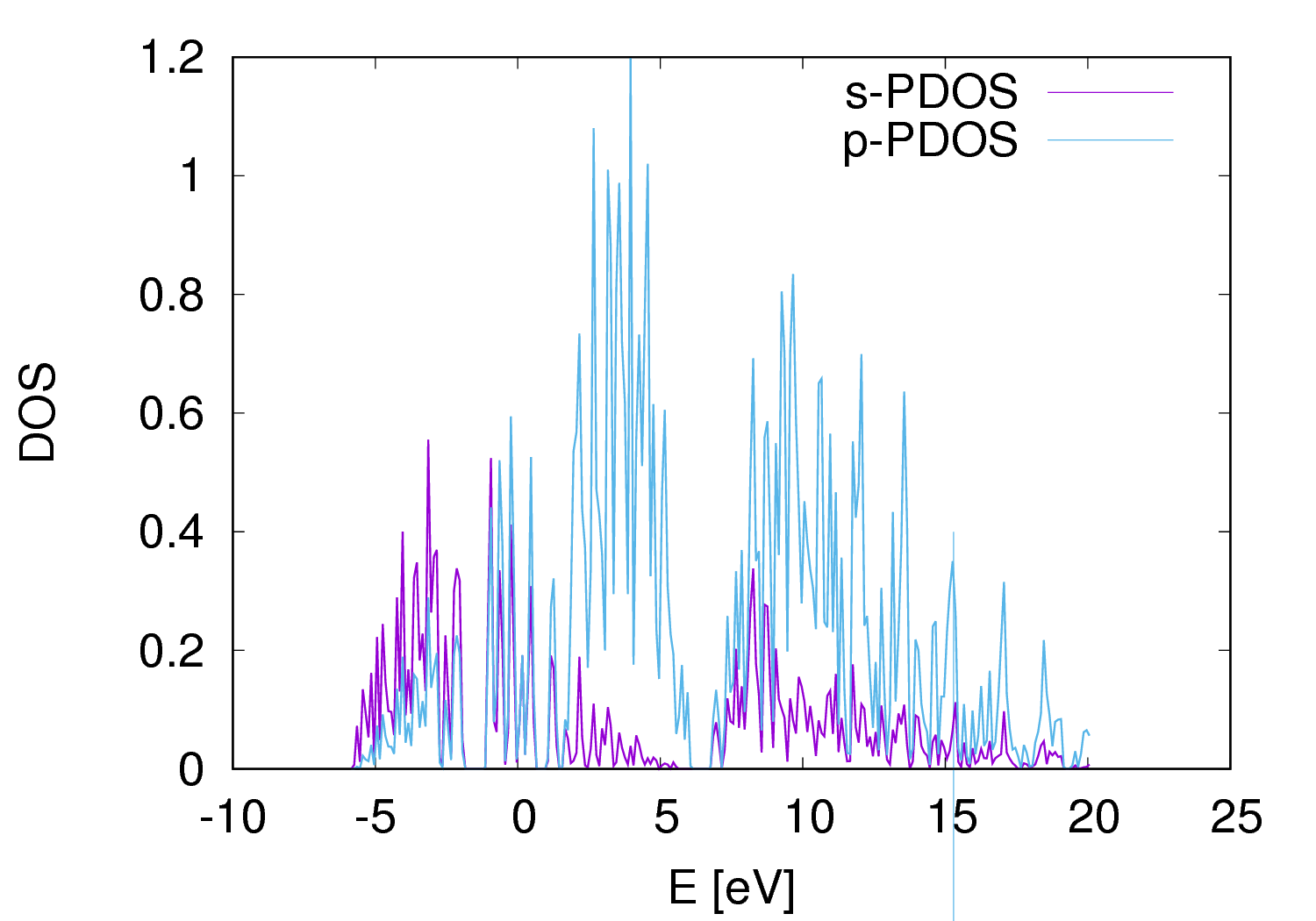
FIG.4: Partial density of state (pDoS) plot of silicon crystal. As seen the near band edge states are dominated by Si-p orbitals. F. Band Structure Calculation
1. Prepare an input file si-bands.in for nscf calculation along the high symmetry paths in the first Brillouin zone. Execute the calculation with
2. Collect the band results for plotting using the code bands.x, which reads data file(s), extracts eigenvalues, regroups them into bands. The output is written to a file that can be directly read and converted to plottable format. A shell script plot_bandstr.sh is prepared to automate this plotting process. 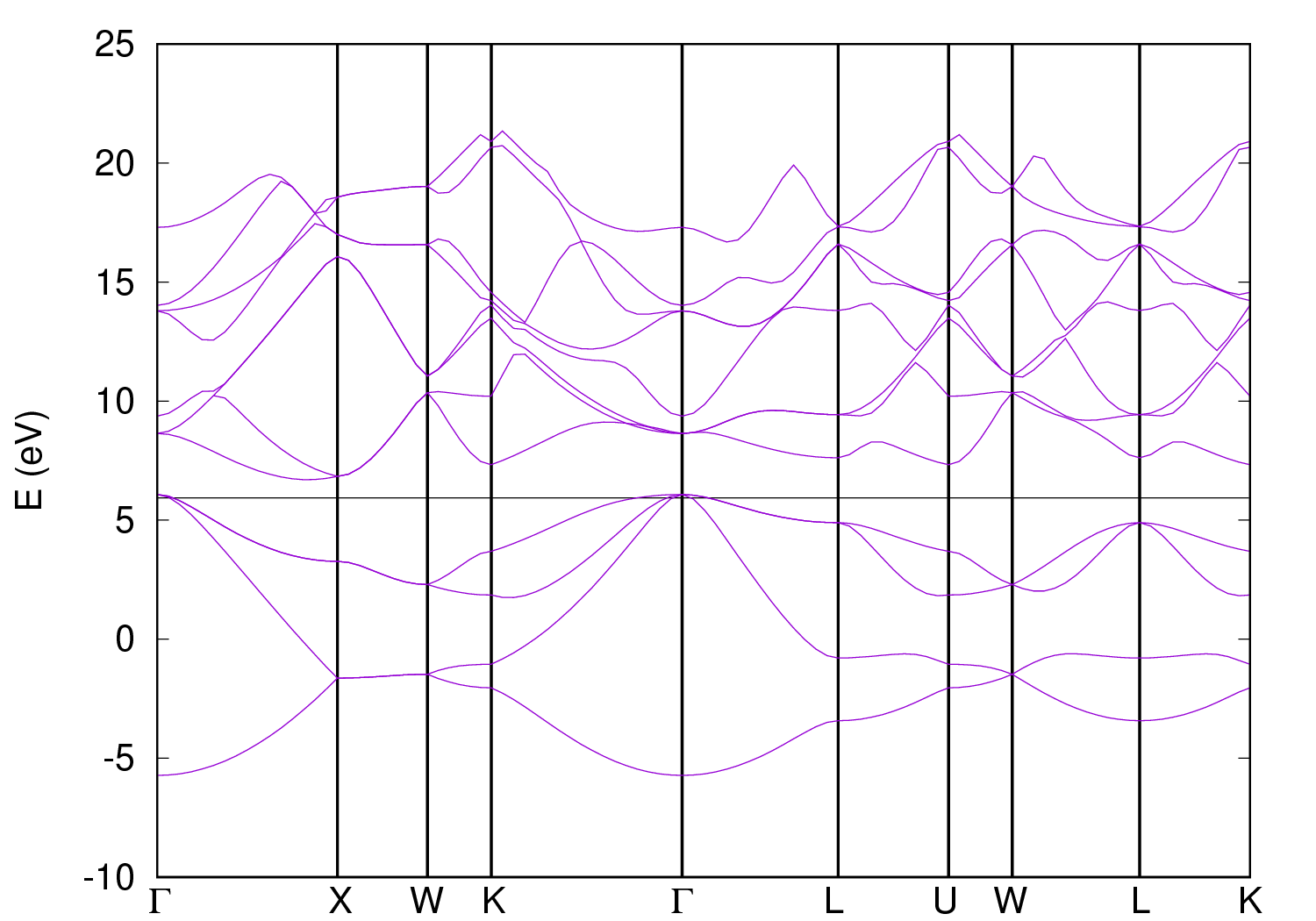
FIG.5: Bandstructure of silicon crystal. Metallic Crystal: AluminumA. SCF Run for visualizing charge density distribution
1. Prepare a shell script run-al.scf.sh for scf calculation. Execute the code with 
FIG.6: Charge density plot on (110) plane of aluminum crystal. B. Conduct a nscf Run for Analyzing Density of State
1. Prepare a shell script run-al.nscf.sh for nscf calculation on 8x8x8 k-mesh. Execute the code with 
FIG.7: Plot of density of state of Al crystal.
4. Producing Partial DoS plot with run-al.pdos.sh and execute the code. C. Band Structure Calculation
1. Prepare an input file al-bands.in for nscf calculation along the high symmetry paths in the first Brillouin zone. Execute the calculation with
2. Collect the band results for plotting using the code bands.x, which reads data file(s), extracts eigenvalues, regroups them into bands. The output is written to a file that can be directly read and converted to plottable format. A shell script plot_bandstr.sh is prepared to automate this plotting process. 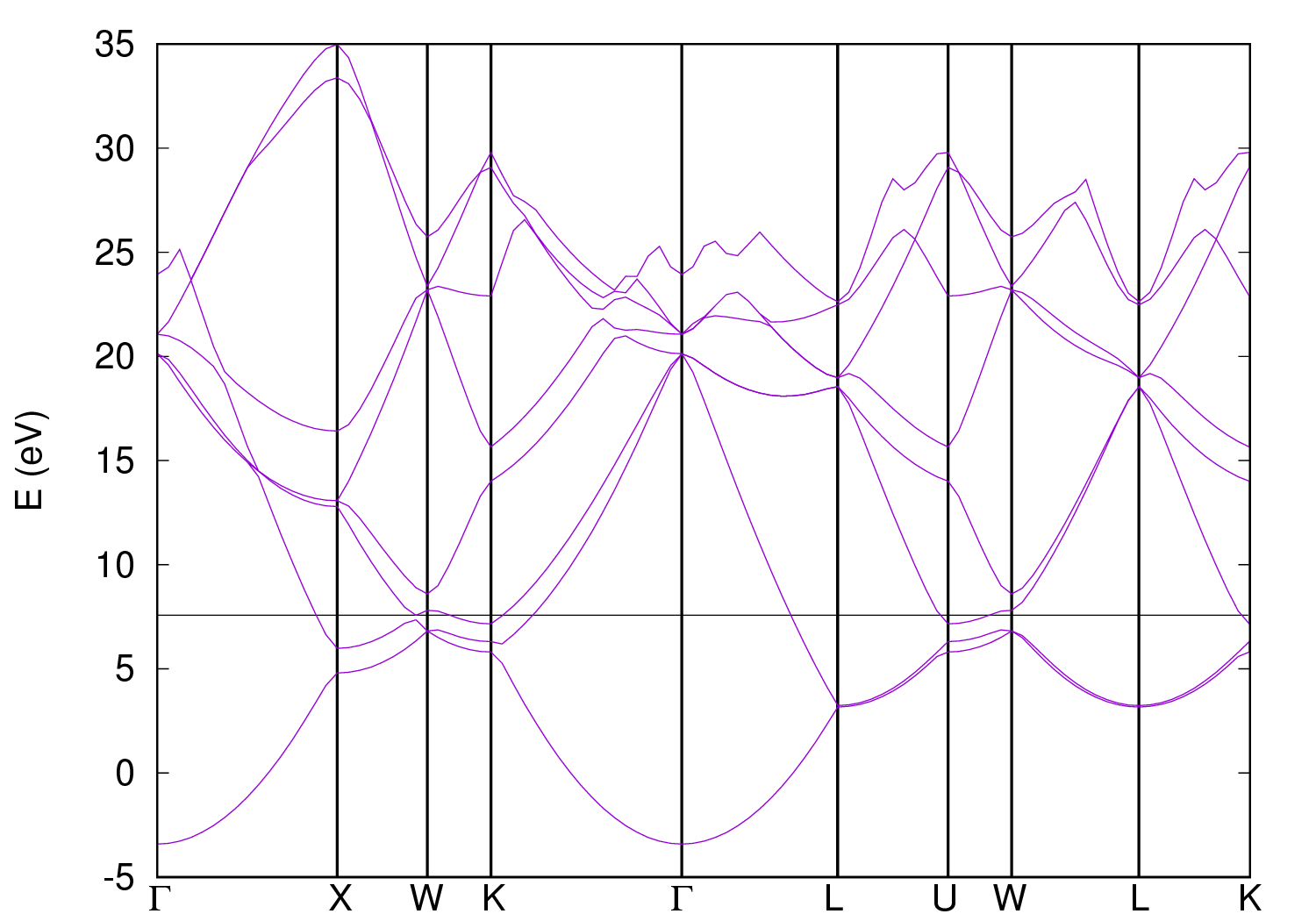
FIG.8: Bandstructure of Al. Structural relaxation on Al(001) surfaceThis example demonstrates how to prepare a model for studying surface adsorption.
A. SCF without Structural Relaxation B. SCF with Structural Relaxation
1. Structural relaxation of Al(001) slab can be done by specifying the 'calculation' flag with 'relax', which allows atoms to relax in the supercell. Modify the input file to perform a structural relaxation with the BFGS method and save the new input as al001-BFGS.in |
Oxygen Adsorbed on Al(100)A longstanding problem on ab initio calculation is to visualize the charge redistribution and transfer caused by an adsorbate on a metal surface. In this example, I will show how to do it. A. O-Al(100) System
1. For the system of oxygen atom adsorbed on Al(100) surface, we need to run the scf calculation of oxygen atom, Al(100) slab, and O-Al(100). A shell script run-Al-O-OAl.sh was prepared to automate the calculations: 
FIG.1: Plot of charge difference of O-Al(100). |
ESM Model for ElectrochemistryElectrochemistry is an extremely important topics in energy generation and storage. In this section, a case study is presented to show the simulation steps with the Effective Screening Medium (ESM) model, which can be used to yield the total energy, charge density, force, and potential of a polarized or charged medium. ESM screens the electronic charge of a polarized/charged medium along one (perpendicular to the interfaces) direction by introducing a classical charge model and a local relative permittivity into the first-principles calculation framework. This permits calculations using open boundary conditions (OBC). The method was described in detail in M. Otani and O. Sugino, "First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach," PRB 73, 115407 (2006).
In addition to 'pbc' (ordinary periodic boundary conditions with ESM disabled), the code allows three different sets of boundary conditions perpendicular to the polarized medium: Simulation Steps
1. A shell script runESM.sh can be downloaded here to facilitate the calculation:
2. Analyze the results: |
FerromagnetismIn this section, I will describe the simulation procedures for ferromagnetic crystal. BCC iron was chosen for this case study. First, we will compute total energy and magnetization versus the lattice parameter with reveal the most stable dimension of the unit cell. By using this unit cell dimension, we will compute the spin-polarized band structure of Fe.bccFM (pw.x + bands.x). After that, we compute the spin-polarized density of states of Fe.bccFM(pw.x + projwfc.x). Finally, fixed spin moment (fsm) calculation was performed at various lattice parameters of fcc Fe to display the difference with that of bcc Fe. A. Total Energy and Magnetization of BCC Iron Crystal versus Lattice Constant
To save your effort, you can download a shell script "pw.alat.sh" here to automate the simulation work. This script can run pw.x for several lattice parameters (the output files are stored in a single file by using the double redirection >>). A simple bash command (grep+awk) is searching for the total energy and total magnetization of the system. Run the shell script as follows:  
FIG.1: Total energy and magnetization of ferromagnetic bcc iron crystal plotted as a function of lattice constant (Bohr). B. Compute the spin-polarized band structure of Fe.bccFM
The band structure is performed in three steps: 
FIG.2: Spin polarized Bandstructure of Fe.bccFM. C. Spin Polarized Density of States of Fe.bccFM
The DOS can be calculated in three steps:
Compare the atomic-projected magnetization (output of projwfc.x) and the magnetization integrated in a sphere (output of pw.x). 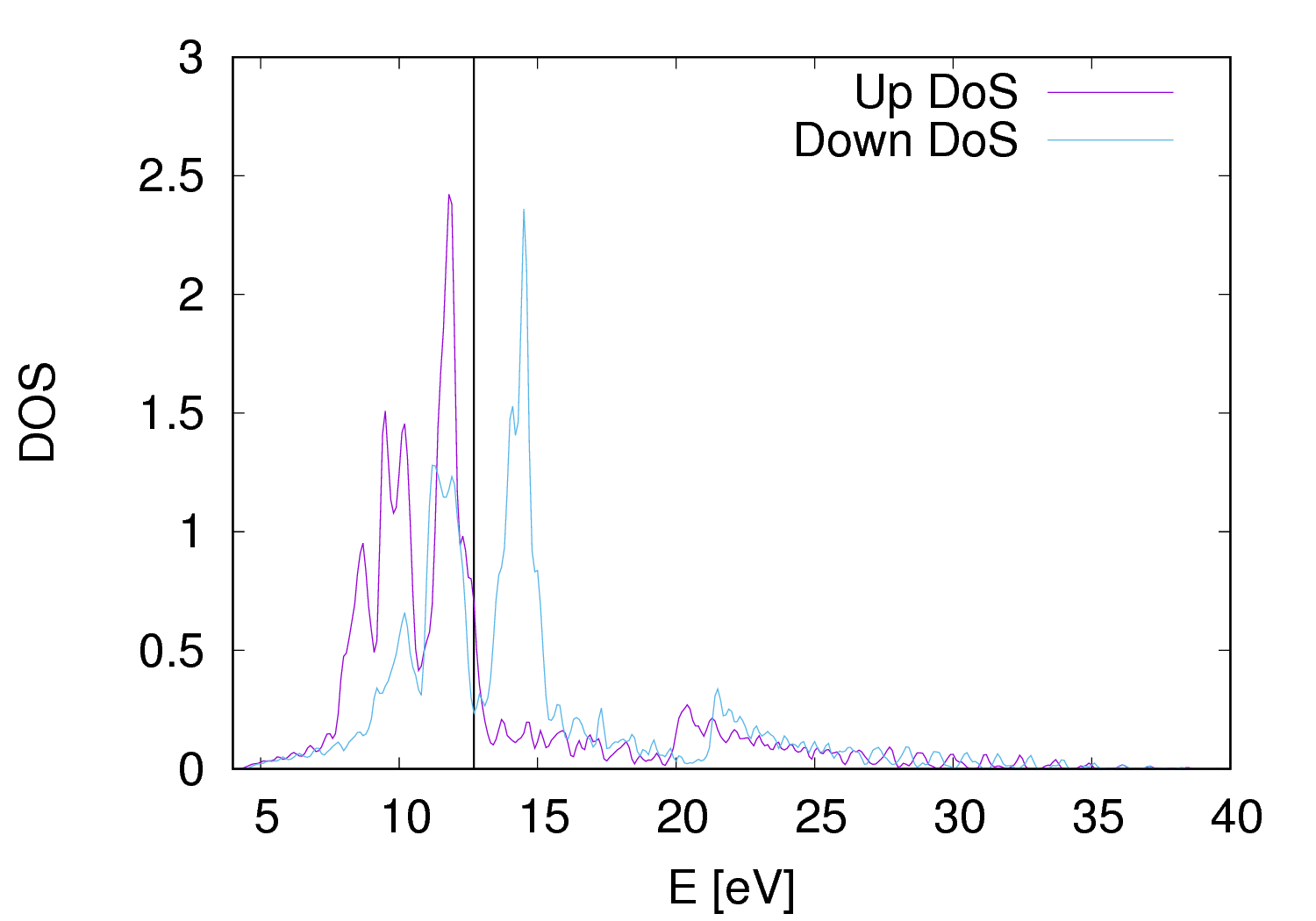 
FIG.3: Plots of density of state (Left) and partial density of states (Right) of Fe.bccFM crystal. The vertical line at 12.5 eV marks the position of the Fermi level. D. Fixed spin moment (fsm) calculation at various lattice parameters of Fe.fcc.FM
The structure considered is Fe.fcc, which has a more complex magetic behavior. In particular, two magnetic solution (Low spin and High Spin) are coexisting in a range of lattice parameters. 
FIG.4: Total energy vs spontaneous magnetization of Fe.fccFM crystal. |
Ferroelectricity: Berry Phase for Electric PolarizationIn this study, we will learn how to use Berry phase formalism of electric polarization to calculate the electric and ionic polarizations for symmetric (A) and asymetric (B) BaTiO3 crystals. To probe into this issue deeply, the effective Born charge generated by displacing the central Ti atom from its symmetric lattice position (C) is also calculated to reveal the origin of the electric polarizations. A. Calculate Polarization via Berry Phase for Symmetric Tetragonal BaTiO3
1. Do a pw scf calculation of total energy on the equilibrium structure with well converged k-mesh and ecutwfc. Prepare the input file "sy.scf.in" with ibrav= 6, K_POINTS=5 5 5. B. Electric and Ionic Polarizations of BaTiO3 with Asymmetric Lattice StructureOptimize the structure of tetragonal BaTiO3 with displaced Ti atom, and then calculate the polarization via Berry phase.
1. Do a pw relax calculation. Prepare the input file "relax.in" (calculation = 'relax', tprnfor = .true., ibrav= 6, with a displaced Ti atom along z).
8. Spontaneous polarization of tetragonal BaTiO3 C. Effective Born Charge Generated by Displaced Ti Atom in a Cubic BaTiO3
1. Run nscf calculation with input file scf.in (ibrav= 1 with the Ti atom displaced by 0.01a along +z direction) |
Phonon Dynamics and Thermal Conductivity of Lattice.
|