|
|
|
This webpage aims to serve as an archive to help me do MD simulations. Detailed procedures of preparing model and performing MD are illustrated. Three examples are included: adenylate kinase (AdK) for MD and well-tempered metaD, liganded protein for modelling drug-protein complex, and a prototypical example of a receptor protein in a lipid bilayer. I have gone through every step to make sure it works. I hope these examples can be modified to fit to your need. |
Adenylate kinase in an aqueous solutionAdenylate kinase (AdK) is the key enzyme in maintaining the cellular energy homeostasis. AdK comprises three relatively rigid domains, central core (CORE), an AMP-binding domain (AMPbd), and a lid-shaped ATP-binding domain (ATPlid). During the catalytic cycle of AdK, the largest conformational changes occur in the ATPlid and the AMPbd domains, while the CORE remains relatively unchanged. During the catalytic reaction, several metastable configurations exist to bridge the open and closed states of the enzyme. Here we describe the molecular dynamic (MD) simultion procedure of AdK with Gromacs 5.1.2 and Plumed 2.2. First we will show how to prepare the files needed for the simulation and how to analyze the MD trajectories. We will also show the detailed step to extract free energy profile with metadynamics by combining Gromacs and Plumed. Regular Molecular Dynamic SimulationA. Prepare the Structural and Topological Files of the Protein
In the folder of working space, I will create an AdK folder for this simulation. In the AdK folder, for the concern of organized data, it is helpful to create some other subfolders: coord (pdb, etc.), mdp (input parameters for md run), md (md run), WTmetaD, etc.
Next, we have to fix the errors or missing residues of the pdb file using the handy online tool "pdbfixer": On the "Welcome To PDBFixer!" page, input your 3adk.pdb file and hit "Analyze file" button. This PDB file contains 2 chains. Select chain #1 (Protein, 195 residues) to be included. Hit "Continue" button. Delete all heterogens atoms (except amino acid residues and nuclei acids). Do not add missing Hydrogens. Deselect "Add water" and then hit "Continue" button. Save the fixed file!
Next, we will prepare the structure, topology (.top) and relevant topology include file (.itp) using the pdb file. Three new files will be generated: adk.gro, adk.top, and adk_posre.itp from pdb2gmx. B. Prepare a Simulation Box, Solvate the Protein and Neutralize the Simulation System
Generate a simulation box and center (-c) the protein in the box using editconf, which needs the processed protein structural file .gro. Allow at least 1.0 nm (-d 1.0) from the rim of the protein to the boundary of the cubic box (-bt cubic).
The output of pdb2gmx told us that the protein has a net charge of -4e. To neutralize the charges, we prepare the needed ions topology file (-o .tpr) by using grompp with parameters included with (-f ions.mdp), (-c) coordinates and (-p) topology information. C. Relax Solvent Configuration with Energy Minimization
This will relax the solvent structure and ions through an energy minimization process while fix the protein position. The following files will be produced: 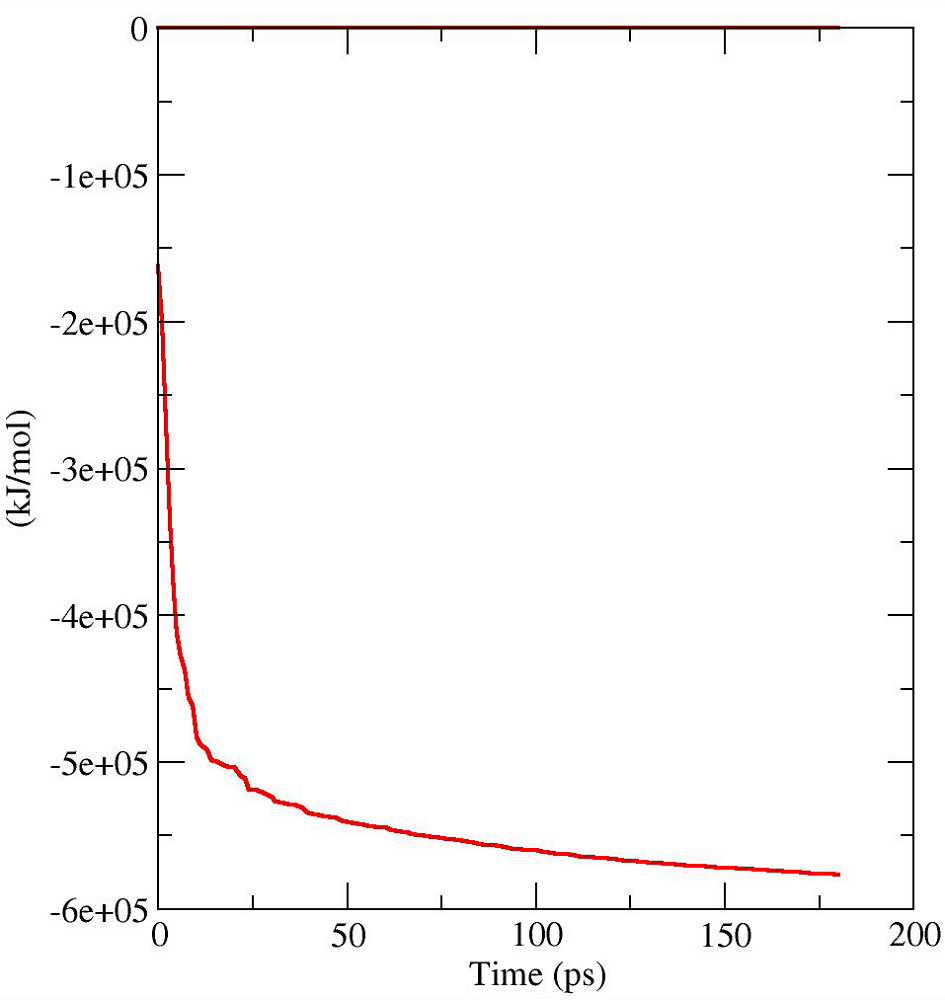
FIG.1: Time course of potential energy in an energy minimization run. D. Equilibrate the System
Using the energy-minimization structure (-c) adk_em.pdb as an input, first perform a NVT MD run to equilibrate the system at a given temperature. 
FIG.2: Time course of system temperature in a NVT run.
Check that the system was brought up to 300K. 
FIG.3: Time course of total density in a NPT run. Check that the pressure fluctuates randomly at 1 bar; the density fluctuates weakly at 1017 kg/m3 E. Production MD Run
Run a 2-ns production MD using an input structure from adk_npt.gro and full precision trajectory file (-t .cpt) from npt run. Use this md.mdp file to prepare .tpr file for a production MD run. F. Trajectory Analysis with bio3D
I. Prepare structural (adk_md.tpr) and trajectory files (adk_md.xtc) for bio3d analysis
II. Trajectory Analysis with bio3d---Launch and Data Input
III. Trajectory Analysis with bio3d---RMSD 
FIG.4: RMSD of AdK structure plotted as a function of frame number (with a frame time of 4 ps).
> rf<-rmsf(protdcd) 
FIG.5: RMSF Analysis revealing the fluctuating residues of the protein.
> pc<-pca.xyz(protdcd) 
FIG.6: PCA Analysis revealing the most significant collective motions. The first three PCAs contribute 52% fluctuation of the protein.
> plot.bio3d(pc$au[,1],ylab="PC1(Blue), PC2(Red)",xlab="Residue Position", typ="l",col="blue",lty=1,lwd=3) 
FIG.7: Two most significant PCA components are decomposed into the RMSFs of each residue.
IV. Trajectory Analysis with bio3d---Normal Mode Analysis (NMA) 
FIG. 8: Graph (deduced from normal mode analysis) showing coupling between different domains (nmp, lid and core) of AdK. G. Determine the rigid domains of 3ADK Protein Structure
Go to the website of "http://pisqrd.escience-lab.org/" 
FIG. 9: Model showing three different domains (core: blue and gray, lid: green, and nmp: red) of AdK. Well-Tempered Metadynamics SimulationA. Prepare the Structural/Topological Files and Perform a Unbiased MD Run
Go to the folder of Adk working space. Perform a unbiased MD run. B. WTmetaD
In the WTmetaD folder, perform a well-tempered metadynamics MD run using this plumed.dat file. 
FIG. 10: Hills added as WTmetaD of AdK.
2. Free Energy Mapping 
FIG. 11: Free energy surface of AdK on theta1 and theta2
3. Calculate one-dimensional free energies from the two-dimensional metadynamics simulation  
FIG. 12: Free energy of AdK projection on (left) theta1 and (right) theta2. |
Liganded ProteinA longstanding goal in structure based drug discovery is to predict ligand binding free energies accurately. In this regards, T4 Lysozyme complexed with 2-propylphenol serves as a prototypical example for calculating the absolute binding free energy of different compounds to the hydrophobic model binding site. This example aims to illustrate how to prepare a complex model of a protein (T4 Lysozyme) with a ligand (2-propylphenol) for MD simulation. A. Prepare the Structure and Topological File of the Complex
In the folder of working space, first I created an AdK folder for this simulation. In this AdK folder, I also created some subfolders to organize data: coord (pdb, etc.), mdp (input parameters for md run), and md (md run), etc.
Next, we select the protein and fix the missing residues of the pdb file using the handy online tool "pdbfixer": On the "Welcome To PDBFixer!" page, input your 3HTB.pdb file and hit "Analyze file" button. This PDB file contains 1 protein chain and JZ4 ligand. Select chain #1 (the protein) to be included. Hit "Continue" button. Delete all heterogens atoms (except amino acid residues and nuclei acids). Do not add missing Hydrogens. Deselect "Add water" and then hit "Continue" button. Save the fixed file as Prot.pdb!
Next, we will prepare the structure and topology of JZ4 compound. However, some hydrogen atoms are missing in JZ4.pdb, so we use obabel to insert the missing H atoms (-h) to JZ4.pdb (-ipdb) and then output as mol2 format (-omol2):
Prepare the topology .top and coordinates file conf.pdb of the protein (without ligand). B. Prepare the Complex
$ cd ../top
If there are crystal water molecules in the structure, there will be a line indicating the number of solvent molecules (SOL) in the topology. In this case, the line above should be inserted before any SOL and ions lines. See below for an example Prot.top file. Name the new topol file as "complex.top" and stores it in the folder of "./top/". C. Gromacs Setup Procedure
Now we are back to the usual GROMACS setup procedure. Up one level to the folder of ./LigProtein: 
FIG.2: Model of JZ4(red)-T4 lysozyme (coloring by residue type) complex . D. Relax SOL/Ions Positions with Energy Minimization
Now we first to relax the solvent molecules and ions with a fixed protein using energy minimization procedure: E. Equilibrate the System
Applying restraints to the ligand. In the nvt.mdp, set F. Perform a Production MD
Run a 1-ns production MD simulation |
EGFR Dimer in a Lipid BilayerThe epidermal growth factor receptor (EGFR) is a transmembrane protein, which contains an extracellular binding domain, a single transmembrane spanning domain, and a cytoplasmic tyrosine kinase domain. Ligands bind to the extracellular domain of EGFR, stimulating conformational changes that support receptor dimerization. Receptor dimerization leads to the activation of the intracellular tyrosine kinase domain and phosphorylates the dimerization partner on specific tyrosine residues. The phosphorylated tyrosine residues then function as docking sites for adaptor proteins, which further activate intracellular signaling cascades. This example aims to illustrate how to prepare a model of a membrane protein (EGFR dimer) embedded in a lipid bilayer for MD simulation. To simplify the simulation only the transmembrane spanning domain of EGFR is included. [I]. Prepare Gromacs Forcefield for Lipids
The Gromacs pdb2gmx engine can only process proteins, nucleic acids, ions and a few co-factors. We need to manually add the topology of POPC to the force-field topology files. However, if using CHARMM36 FF, then no need to modify the contents of the bonded.itp and nonbonded.itp files, as CHARMM36 already includes parameters for some of the commonly used lipid molecules like POPC. [II]. Prepare folders and relevant Gromacs files needed MD
Using the following cmdline scripts: [III]. Assemble protein in lipid bilayer system
1. Insert hEGFR2 into the lipid bilayer 
FIG.1: Model of hEGFR dimer in a POPC bilayer. Here only transmembrane helix and cytoplasma segment of EGFR are displayed.
3. Embedding the Protein in POPC Bilayer with InflateGRO2 [IV]. Solvate the membrane protein system with water
First, we have to avoid potential contact between solvent molecules with lipid and protein as the system is solvated. This can be done as follow: [IV]. Neutralize the membrane protein system
Use this ion.mdp file to prepare .tpr file for genion. [V]. Relax Solvent Configuration by Energy Minimization
Use this em.mdp file to prepare energy minimization run. [VI]. Equilibrate the System
For a typical MD simulation of membrane protein, it is useful to create special index groups consisting of (solvent + ions) and (protein + lipids) for better equilibration: [VII]. Production MD
Use this md.mdp file to prepare a production MD run. |